Al margen un sello con el Escudo Nacional, que dice: Estados Unidos Mexicanos.- Secretaría de Salud.
MIKEL ANDONI ARRIOLA PEÑALOSA, Comisionado Federal para la Protección contra Riesgos Sanitarios y Presidente del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, con fundamento en los Arts. 39, de la Ley Orgánica de la Administración Pública Federal; 4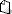 , de la LFPA; 3, fraccs. XXIV y XXV, 13, apartado A, fracs. I, II y IX, 17 bis, fraccs III y VI, 100, 102, 107, 195, 214, 222, 222 Bis, 257, 258, 259, 260, fracc. I, 376, 376 Bis, y 391 Bis, de la Ley General de Salud; 38, fracc. II, 40, fraccs. I y XI, 41, 43 y 47, fracc. IV, de la LFSMN; 8, 81, 153, 161, 161-bis, 177 a 177 Bis 5 y 186, del Reglamento de Insumos para la Salud; 28, del Reglamento de la LFSMN; 36 y 37, del Reglamento Interior de la SS; 3, fraccs. I literal b y II, 10, fraccs. IV y VIII, del Reglamento de la COFEPRIS, y
, de la LFPA; 3, fraccs. XXIV y XXV, 13, apartado A, fracs. I, II y IX, 17 bis, fraccs III y VI, 100, 102, 107, 195, 214, 222, 222 Bis, 257, 258, 259, 260, fracc. I, 376, 376 Bis, y 391 Bis, de la Ley General de Salud; 38, fracc. II, 40, fraccs. I y XI, 41, 43 y 47, fracc. IV, de la LFSMN; 8, 81, 153, 161, 161-bis, 177 a 177 Bis 5 y 186, del Reglamento de Insumos para la Salud; 28, del Reglamento de la LFSMN; 36 y 37, del Reglamento Interior de la SS; 3, fraccs. I literal b y II, 10, fraccs. IV y VIII, del Reglamento de la COFEPRIS, y
CONSIDERANDO
Que en cumplimiento a lo previsto en el Art. 46, fracción I, de la LFSMN, el 10 de abril de 2013, el Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, aprobó el anteproyecto de la presente Norma;
Que con fecha 6 de mayo de 2013 , en cumplimiento a lo previsto en el Art. 47, fracción I, de la LFSMN, se publicó en el DOF, el proyecto de la presente Norma, a efecto de que dentro de los 60 días naturales siguientes al de su publicación, los interesados presentaran sus comentarios al Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario;
Que con fecha previa, fueron publicadas en el DOF, las respuestas a los comentarios recibidos por el mencionado Comité, en términos del Art. 47, fracción III, de la LFSMN, y
Que en atención a las anteriores consideraciones, contando con la aprobación del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, he tenido a bien expedir y ordenar la publicación en el DOF, de la siguiente
NORMA OFICIAL MEXICANA NOM-257-SSA1-2014, EN MATERIA DE MEDICAMENTOS BIOTECNOLOGICOS
0. Introducción
Mediante el Decreto por el que se adiciona el Art. 222 Bis, publicado en el DOF el 11 de junio de 2009, el cual entró en vigor el 10 de septiembre de 2009, se estableció que, tratándose de medicamentos biotecnológicos, sólo puede y debe haber en México medicamentos biotecnológicos innovadores y medicamentos biotecnológicos biocomparables.
El Art. 222 Bis, de la LGS establece el régimen legal de los medicamentos biotecnológicos en México y prevé que todos los medicamentos biotecnológicos deben obtener el registro sanitario, siempre y cuando cumplan con los requisitos y pruebas que demuestren la calidad, seguridad y eficacia del producto. Asimismo, se señala que los solicitantes de medicamentos biotecnológicos biocomparables que sustenten su solicitud en un medicamento biotecnológico de referencia, deberán presentar los estudios de biocomparabilidad.
El 19 de octubre de 2011, se publicó en el DOF el Decreto por el que se reforman y adicionan diversas disposiciones del RIS, el cual entró en vigor el 17 de abril de 2012, estableció los requerimientos que permiten verificar la calidad, seguridad y eficacia de los medicamentos biotecnológicos innovadores y biocomparables, en función de las características y complejidad de su propia naturaleza.
Desde el 2012 las disposiciones normativas complementarias en materia de medicamentos biotecnológicos se han ido configurando en diversas NOM´s de acuerdo a su temática regulatoria, definida, en su momento, mediante la NOM-EM-001-SSA1-2012, Medicamentos biotecnológicos y sus biofármacos. Buenas prácticas de fabricación (Sin embargo, a partir del 23 de octubre de 2013 quedó cancelada la NOM-EM-001-SSA1-2012, debido a que las circunstancias que motivaron su expedición ya fueron mitigadas con esta NOM, Aviso DOF 22/X/2013). Características técnicas y científicas que deben cumplir éstos para demostrar su seguridad, eficacia y calidad. Etiquetado. Requisitos para realizar los estudios de biocomparabilidad y farmacovigilancia, publicada en el DOF el 20 de septiembre de 2012. Dicha Norma de Emergencia, estuvo vigente hasta la publicación en el DOF de su aviso de cancelación, el 22 de octubre de 2013.
Como parte de la configuración de las disposiciones normativas sobre medicamentos biotecnológicos el 20 de septiembre de 2013, se publicó en el DOF la NOM-177-SSA1-2013, Que establece las pruebas y procedimientos para demostrar que un medicamento es intercambiable. Requisitos a que deben sujetarse los Terceros Autorizados que realicen las pruebas de intercambiabilidad. Requisitos para realizar los estudios de biocomparabilidad. Requisitos a que deben sujetarse los Terceros Autorizados, Centros de Investigación o Instituciones Hospitalarias que realicen las pruebas de biocomparabilidad.
Antes de la existencia del marco normativo de los medicamentos biotecnológicos descrito anteriormente no se contaba con definiciones, clasificaciones, así como tampoco con requisitos técnicos y científicos para este tipo de medicamentos. Los constantes avances científicos y tecnológicos, así como la experiencia en política farmacéutica y regulatoria de organismos internacionales como son la OMS y la Agencia Europea de Medicamentos (EMA) han permitido a México avanzar en los aspectos normativos necesarios de evaluación, aprobación y control de estos medicamentos. En este sentido el marco normativo de los medicamentos biotecnológicos requiere de una constante revisión y actualización.
Además de responder a la necesidad constante de fortalecimiento normativo que demandan estos medicamentos en sus procesos de autorización sanitaria, es necesario que los medicamentos de origen biotecnológico que fueron autorizados antes de la reforma legal y que aún se encuentran en el mercado, ingresen en un proceso de revisión de sus registros sanitarios en apego al Art. 157, del Reglamento de Insumos para la Salud, a fin de que cumplan con los requisitos que establece la Ley General de Salud.
Por lo anterior es indispensable contar con una NOM en materia de medicamentos biotecnológicos que fortalezca su marco regulatorio y establezca una configuración completa, actualizada y ordenada de disposiciones normativas para la autorización sanitaria de estos medicamentos.
1. Objetivo
Esta Norma tiene por objeto establecer:
1.1 Las directrices generales de operación para la evaluación de la información técnica y científica presentada durante el proceso de la solicitud de registro de medicamentos biotecnológicos.
1.2. Los criterios por los cuales la Secretaria llevará a cabo el proceso de regularización de los medicamentos biotecnológicos.
1.3 Las especificaciones generales para el control de la fabricación de los medicamentos biotecnológicos.
1.4 El procedimiento para la autorización de protocolos de ensayos clínicos de medicamentos biotecnológicos.
1.5 Las especificaciones que deben cumplir los medicamentos biotecnológicos para ser reconocidos como medicamentos biotecnológicos de referencia.
2. Campo de aplicación
2.1 Esta Norma es de observancia obligatoria en todo el territorio nacional, para los solicitantes de registro sanitario y prórroga de los medicamentos biotecnológicos.
3. Referencias
Para la correcta aplicación de esta Norma, es necesario consultar las siguientes NOM´s o las que las sustituyan:
3.1 NOM-012-SSA3-2012, Que establece los criterios para la ejecución de proyectos de investigación para la salud en seres humanos.
3.2 NOM-059-SSA1-2013 (Actual NOM-059-SSA1-2015) Buenas prácticas de fabricación de medicamentos.
3.3 NOM-072-SSA1-2012 Etiquetado de medicamentos y de remedios herbolarios.
3.4 NOM-073-SSA1-2005 Estabilidad de fármacos y medicamentos.
3.5 NOM-164-SSA1-2013 Buenas prácticas de fabricación para fármacos.
3.6 NOM-177-SSA1-2013 Que establece las pruebas y procedimientos para demostrar que un medicamento es intercambiable. Requisitos a que deben sujetarse los Terceros Autorizados que realicen las pruebas de intercambiabilidad. Requisitos para realizar los estudios de biocomparabilidad. Requisitos a que deben sujetarse los Terceros Autorizados, Centros de Investigación o Instituciones Hospitalarias que realicen las pruebas de biocomparabilidad.
3.7 NOM-220-SSA1-2012 Instalación y operación de la farmacovigilancia.
4. Símbolos y abreviaturas
Cuando en esta Norma se haga referencia a las siguientes abreviaturas, se entenderá:
4.1 CMN Comité de Moléculas Nuevas.
4.2 COFEPRIS Comisión Federal para la Protección contra Riesgos Sanitarios.
4.3 DOF Diario Oficial de la Federación.
4.4 LGS Ley General de Salud.
4.5 OMS Organización Mundial de la Salud.
4.6 RIS Reglamento de Insumos para la Salud.
4.7 SEPB Subcomité de Evaluación de Productos Biotecnológicos.
4.8 Secretaría Secretaría de Salud.
5. Directrices generales de operación para la evaluación de la información técnica y científica presentada durante el proceso de la solicitud de registro de medicamentos biotecnológicos
5.1 Generalidades.
5.1.1 La COFEPRIS cuenta con un CMN el cual su vez tiene un SEPB que está integrado por especialistas y científicos en materia de biotecnología.
5.1.2 Todos los medicamentos biotecnológicos innovadores, deberán presentarse para ser evaluados ante el CMN y deberán ser estudiados por el SEPB previamente al sometimiento de la solicitud de registro sanitario, para determinar si existen elementos técnicos y científicos para demostrar su seguridad, calidad y eficacia.
5.1.3 La Secretaría, con base en la opinión del CMN, previa consulta que éste realice al SEPB, determinará las pruebas de biocomparabilidad que permitan la autorización de las indicaciones terapéuticas a los medicamentos biotecnológicos biocomparables.
5.1.4 El SEPB para la evaluación de la información técnica y científica presentada durante el proceso de la solicitud de registro y prórroga de medicamentos biotecnológicos aplicará, conforme al Reglamento Interior del CMN.
5.1.5 El SEPB de conformidad con su reglamento, podrá evaluar cualquier información técnica y científica, y emitirá una opinión relacionada con:
5.1.5.1. Nuevos medicamentos biotecnológicos durante su etapa de investigación y/o desarrollo;
5.1.5.2 Nuevos medicamentos biotecnológicos con sus etapas de investigación y/o desarrollo concluidas;
5.1.5.3 Medicamentos biotecnológicos biocomparables, tanto en etapa de desarrollo como con estudios concluidos;
5.1.5.4 Medicamentos biotecnológicos que ya cuentan con un registro sanitario y que se encuentren en proceso de prórroga;
5.1.5.5 Clasificación de medicamentos biotecnológicos como innovadores o biocomparables, y
5.1.5.6 Definición de medicamentos de referencia.
6. Control de la fabricación de medicamentos biotecnológicos
6.1 El particular deberá someter tanto para un medicamento biotecnológico innovador como para un medicamento biotecnológico biocomparable, el programa de aseguramiento de calidad del producto, que establezca lo siguiente:
6.1.1 Programa de validaciones y/o revalidaciones de los procesos;
6.1.2 Información de los parámetros críticos de validación obtenidos;
6.1.3 Programa de auditorías internas del producto y del proceso;
6.1.4 Para las validaciones y programas de auditorías internas deberán reportar las acciones preventivas y correctivas relacionadas con los parámetros críticos del producto y del proceso;
6.1.5 La información anterior deberá ser reportada anualmente a partir de la fecha de emisión del registro correspondiente y sucesivamente durante la vigencia del mismo, y
6.1.6 La presentación de estos reportes será evaluada al momento del análisis de las solicitudes de prórroga para efectos de la emisión del registro.
7. Autorización de protocolos
7.1 Los estudios de investigación clínica para medicamentos biotecnológicos innovadores y medicamentos biotecnológicos biocomparables seguirán el mismo procedimiento de autorización de cualquier protocolo de investigación clínica. En caso de que el estudio tenga fines de registro, el particular podrá notificarlo por escrito junto con la documentación sometida para autorización del protocolo.
8. Farmacovigilancia de medicamentos biotecnológicos
8.1 La Farmacovigilancia de los medicamentos biotecnológicos deberá realizarse conforme a lo establecido en la NOM, citada en el punto 3.8, del Capítulo de Referencias de esta Norma.
9. Requisitos para el reconocimiento de medicamentos biotecnológicos de referencia
9.1 Para efecto de obtener y mantener el reconocimiento como medicamento biotecnológico de referencia, se deberá cubrir lo siguiente:
9.1.1 Contar con un registro sanitario vigente emitido por la COFEPRIS;
9.1.2 Estar disponible comercialmente en territorio nacional;
9.1.3 En caso de que ya no exista un medicamento local de referencia, un medicamento biotecnológico biocomparable podrá ser considerado como tal, siempre y cuando se haya demostrado la biocomparabilidad respecto al medicamento de referencia vigente al momento de la realización del estudio, y
9.1.4 Dar cumplimiento a lo establecido en la NOM citada en el punto 3.8, del Capítulo de Referencias de esta Norma..
9.2 Los casos no previstos, serán evaluados por la COFEPRIS con apoyo del SEPB.
9.3 La COFEPRIS publicará en su página de internet la lista de medicamentos biotecnológicos de referencia, la cual se actualizará dentro de los 10 días siguientes a la fecha de autorización de un medicamento de referencia.
10. Estudios de biocomparabilidad de medicamentos biotecnológicos
10.1 Los estudios y las pruebas de biocomparabilidad deberán realizarse de acuerdo a lo establecido en la NOM, citada en el punto 3.7, del Capítulo de Referencias de esta Norma.
11. Concordancia con normas internacionales y mexicanas
11.1 Esta Norma no es concordante con normas internacionales ni mexicanas.
12. Bibliografía
12.1 Ley General de Salud .
.
12.2 Ley Federal sobre Metrología y Normalización.
12.3 Reglamento de Insumos para la Salud.
12.4 Reglamento de la Ley Federal sobre Metrología y Normalización.
12.5 Reglamento de la Comisión Federal para la Protección contra Riesgos Sanitarios.
12.6 Farmacopea de los Estados Unidos Mexicanos, 10a. ed. México (2009).
12.7 ISO 31000:2009. Principles and Guidelines on Implementation.
12.8 Organización Mundial de la Salud. Manual de bioseguridad en el laboratorio, tercera edición. OMS, Ginebra, 2005.
12.9 World Health Organization. Quality Assurance of Pharmaceuticals. A compendium of guidelines and related materials, Vol. 2, 2a. Edition, 2007.
12.10 World Health Organization. Guidelines on evaluation of similar Biotherapeutic Products (SBPs), ECBS, October 2009.
12.11 World Health Organization. Proposed WHO Risk Management Plan for dealing with copy biologicals inappropriately licensed as “Biogenerics”. Draft_11 May 2012.
12.12 World Health Organization. Guidelines on the Quality, Safety, and Efficacy of Biotherapeutic Protein Products prepared by Recombinant DNA Technology. Replacement of Annex 3 of WHOTechnical Report Series, No. 814 2013.
12.13 World Health Organization. Regulatory expectations and risk assessment for biotherapeutic products. Scientific Principles to Consider. 2014.
12.14 Colegio Nacional de Químicos Farmacéuticos Biólogos México. Guía de Validación del Método Analítico. CNQFBM, 2002.
12.15 Gary Walsh. Pharmaceutical Biotechnology. Concepts and Applications. Ed. John Wiley and Sons Ltd. 2007.
13. Observancia de la Norma
La vigilancia del cumplimiento de esta Norma, corresponde a la SS, para lo cual su personal realizará las acciones de verificación y demás que resulten necesarias.
14. Vigencia
Esta Norma entrará en vigor a los 60 días naturales posteriores a su publicación en el DOF.
TRANSITORIO
UNICO.- Para el caso de las prórrogas de los registros sanitarios de los medicamentos con registro sanitario emitido antes de entrar en vigor las reformas al RIS publicadas en el DOF el 19 de octubre de 2011, se someterán al siguiente procedimiento:
a) Cada solicitud de registro será evaluada por la COFEPRIS, para lo cual realizará la clasificación del producto como innovador o biocomparable.
b) Para la obtención de la prórroga de su registro, los productos innovadores deberán cumplir los requisitos aplicables del Acuerdo por el que se dan a conocer los trámites y servicios, así como los formatos que aplica la Secretaría de Salud, a través de la COFEPRIS, inscritos en el Registro Federal de Trámites y Servicios de la Comisión Federal de Mejora Regulatoria y no requerirán la opinión del SEPB para la obtención de la prórroga del registro sanitario.
c) En el caso de que se haya solicitado la prórroga del registro como producto biocomparable, el SEPB determinará el medicamento de referencia para cada biofármaco, el cual será notificado al particular para que realice y someta ante el SEPB los estudios correspondientes. En caso de que el particular haya realizado las pruebas de caracterización con un medicamento diferente se someterá esta información y otros elementos técnicos que considere el solicitante a evaluación del SEPB para su opinión.
d) Una vez evaluados los elementos técnicos aportados por el solicitante y con base en la opinión del SEPB, la COFEPRIS podrá otorgar al particular un plazo máximo para que aporte información complementaria, por lo que le notificará la vigencia del registro correspondiente hasta en tanto se emita la resolución definitiva sobre la solicitud de prórroga. La COFEPRIS informará a entidades e instituciones públicas y privadas sobre la vigencia y efectos jurídicos plenos para la comercialización de los registros que se encuentren en el supuesto de la presente disposición transitoria.
e) El titular del registro deberá cumplir las fechas establecidas por la COFEPRIS para completar la información señalada por el SEPB, la cual deberá someter en tiempo y forma ante la autoridad sanitaria, máximo 10 días hábiles posteriores al vencimiento del plazo otorgado. En caso de que la información no haya sido presentada en tiempo y forma la COFEPRIS podrá prevenir al titular del registro sanitario para que ésta sea presentada en un plazo determinado, apercibiéndolo de que en caso de no cumplirla se desechará la solicitud correspondiente.
f) Los criterios referidos en los puntos del presente transitorio, serán aplicables únicamente para los registros cuya última fecha de presentación de solicitud de prórroga, conforme a lo previsto en el artículo 190 bis 6 del RIS, sea a más tardar el 31 de diciembre de 2015.
Sufragio Efectivo. No Reelección.
México, D.F., a 11 de diciembre de 2014.- El Comisionado Federal para la Protección contra Riesgos Sanitarios y Presidente del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, Mikel Andoni Arriola Peñalosa.- Rúbrica.
NOTAS:
Esta NOM se publico el 11/XII/2014(Circular G-37/15) . Su PROY-NOM se publicó el 6/V/2013(Circular G-120/13) y su respuesta a los comentarios el 25/XI/2014.
La COFEMER da a conocer en la liga http://207.248.177.30/regulaciones/scd_expediente_3.asp?ID=02/1198/100413 un anteproyecto de este Proyecto de NOM, el cual presenta una nueva versión y trae cambios sustanciales (Circular T-281/14)